Research
High-throughput genomics
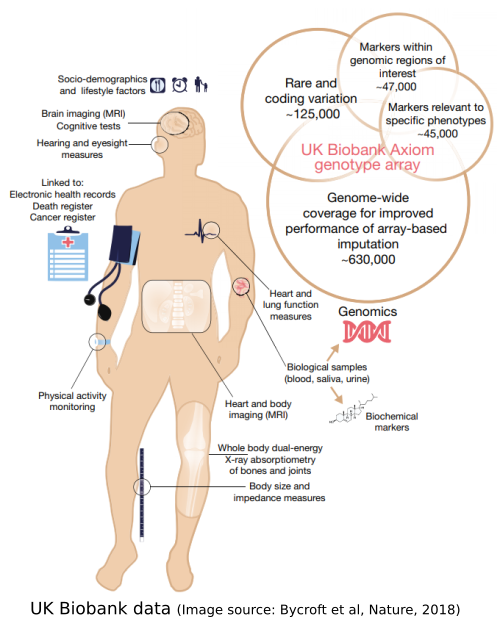
Modern high-throughput sequencing technologies allow us to efficiently make all sorts of measurements genome-wide. These kinds of data have enormous potential for science and medicine, and present a variety of novel statistical challenges. I have a broad interest in genetics and genomics, including genome-wide association studies and CRISPR screens; such applications also motivate my methodological and theoretical research.
High-dimensional variable selection
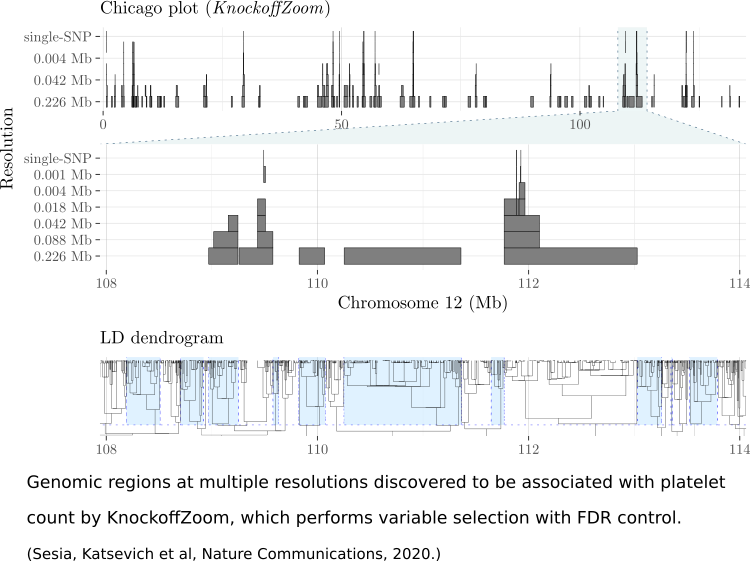
Genome-wide association studies exemplify the situation where we have many input variables (genetic variants) that can potentially affect the outcome variable (disease status), creating a challenging statistical problem. I have developed theory and methods for this problem, in the context of the Model-X framework introduced by Candes et al.
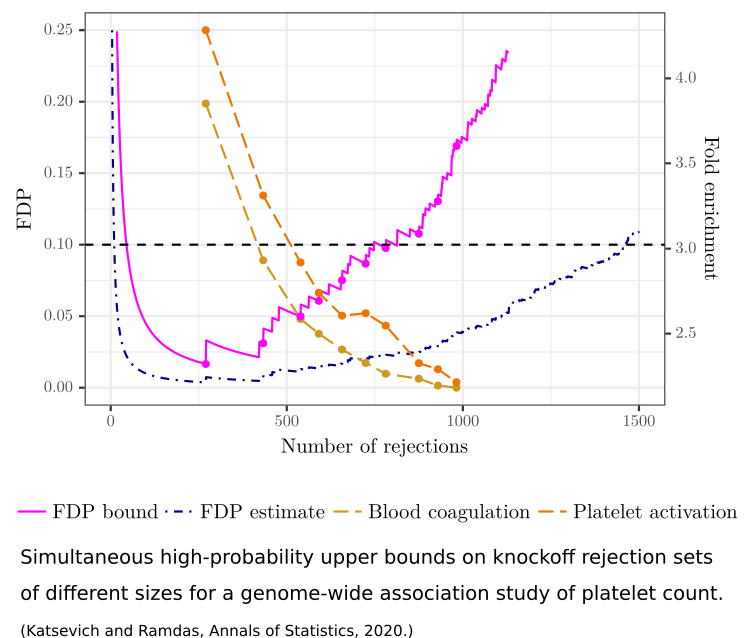
Multiple testing
Multiplicity is a ubiquitous problem in genomics applications, creating various methodological challenges. One challenge is to incorporate the structure of the underlying biology into the multiple testing procedure. For example, the genome has spatial structure, and biological networks have graphical structures. I’ve developed methodology for multiple testing problems involving group structure and graph structure.
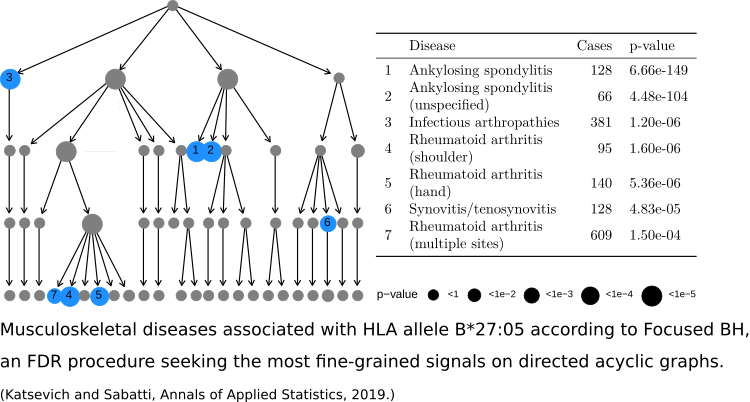
Reconciling exploration and inference
As new technologies facilitate faster and cheaper data collection, scientists increasingly use data for generating (as opposed to confirming) hypotheses. Data exploration is an integral step in hypothesis-generating research, but is at odds with standard statistical significance guarantees. Nevertheless, statistical guarantees are important to ensure the quality of the hypotheses chosen for confirmation. Reconciling exploration with inference is therefore an important statistical challenge. I have proposed a means for doing this in the context of Goeman and Solari’s simultaneous inference paradigm.